[138] Simple Approximation for the Ideal Reference State of Gases Adsorbed on Solid-State Surfaces
J. Ye and D. G. Truhlar
J. Am. Chem. Soc. 144, 12850–12860 (2022)
DOI: 10.1021/jacs.2c04333


view older publications | view newer publications | view published covers
J. Ye and D. G. Truhlar
J. Am. Chem. Soc. 144, 12850–12860 (2022)
DOI: 10.1021/jacs.2c04333
N. Luo, L. Feng, H. Yin, A. Stein, S. Huang, Z. Hou, and D. G. Truhlar
ACS Appl. Mater. Interfaces 14, 29832–29843 (2022)

M. S. Minkara, T. R. Josephson, C. L. Venteicher, B. R. Greenvall, R. K. Lindsey, P. H. Koenig, and J. I. Siepmann
J. Phys. Chem. B 126, 3940–3949 (2022)

D. M. Anstine, D. S. Sholl, J. I. Siepmann, R. Q. Snurr, A. Aspuru-Guzik, C. M. Colina
Curr. Opin. Chem. Eng. 36, 100795 (2022)

S. Pahari, M. Dorneles de Mello, M. S. Shah, T. R. Josephson, L. Ren, H. G. T. Nguyen, R. D. Van Zee, M. Tsapatsis, and J. I. Siepmann
ACS Phys. Chem. Au 2, 79–88 (2022)

D. S. Graham, X. Wen, D. V. Chulhai, and J. D. Goodpaster
J. Chem. Phys. 156, 054112 (2022)
DOI: 10.1063/5.0076493

S. S. Rajasree, J. Yu, S. M. Pratik, X. Li, R. Wang, A. S. Kumbhar, S. Goswami, C. J. Cramer, and P. Deria
J. Am. Chem. Soc. 144, 1396–1406 (2022)
DOI: 10.1021/jacs.1c11979
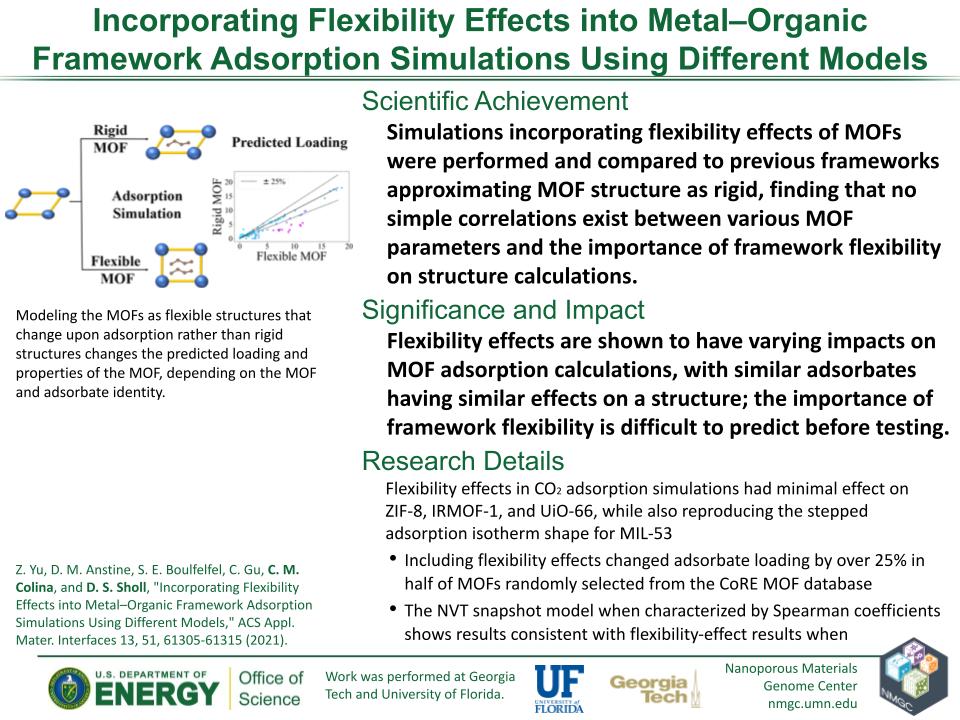
Z. Yu, D. M. Anstine, S. E. Boulfelfel, C. Gu, C. M. Colina, and D. S. Sholl
ACS Appl. Mater. Interfaces 13, 61305–61315 (2021)
W. S. Jeong, C. A. Gaggioli, and L. Gagliardi
J. Chem. Theory Comput. 17, 7518–7530 (2021)

R. J. Drout, M. A. Gaidimas, and O. K. Farha
ACS Appl. Mater. Interfaces 13, 51886–51893 (2021)

X. Yu, S. Choi, D. Tang, A. J. Medford, and D. S. Sholl
J. Phys. Chem. C 125, 18046–18057 (2021)

K. Chen, C. A. Downes, J. D. Goodpaster, and S. C. Marinescu
Inorg. Chem. 60, 11923–11931 (2021)

S. Kanchanakungwankul and D. G. Truhlar
J. Chem. Theory Comput. 17, 4823–4830 (2021)

K. Chen, D. Ray, M. E. Ziebel, C. A. Gaggioli, L. Gagliardi, and S. C. Marinescu
ACS Appl. Mater. Interfaces 13, 34419–34427 (2021)

Y. Sun, R. F. DeJaco, Z. Li, D. Tang, S. Glante, D. S. Sholl, C. M. Colina, R. Q. Snurr, M. Thommes, M. Hartmann, and J. I. Siepmann
Sci. Adv. 7, eabg3983 (2021)

A. G. Demidov, B. L. A. Perera, Mi. E. Fortunato, S. Lin, and C. M. Colina
SoftwareX 15, 100749 (2021)

Z. Li, B. J. Bucior, H. Chen, M. Haranczyk, J. I. Siepmann, and R. Q. Snurr
J. Chem. Phys. 155, 014701 (2021)
DOI: 10.1063/5.0050823

C.-P. Wang, Y. Feng, H. Sun, Y. Wang, J. Yin, Z. Yao, X.-H. Bu, and J. Zhu
ACS Catal. 11, 7132–7143 (2021)

D. M. Anstine and C. M. Colina
Polym. Int 70, 984–989 (2021)
DOI: 10.1002/pi.6124

J. G. Park, B. A. Collins, L. E. Darago, T. Runčevski, M. E. Ziebel, M. L. Aubrey, H. Z. H. Jiang, E. Velasquez, M. A. Green, J. D. Goodpaster, and J. R. Long
Nat. Chem. 13, 594–598 (2021)

A. Tang, X. He, H. Yin, Y. Li, Y. Zhang, S. Huang, and D. G. Truhlar
J. Phys. Chem. C 125, 9679–9687 (2021)

R. Pandharkar, M. R. Hermes, C. J. Cramer, D. G. Truhlar, and L. Gagliardi
J. Chem. Theory Comput. 17, 2843–2851 (2021)

R. Li, S. Alomari, R. Stanton, M. C. Wasson, T. Islamoglu, O. K. Farha, T. M. Holsen, S. M. Thagard, D. J. Trivedi, and M. Wriedt
Chem. Mater. 33, 3276–3285 (2021)

S. Kancharlapalli, A. Gopalan, M. Haranczyk, and R. Q. Snurr
J. Chem. Theory Comput. 17, 3052–3064 (2021)

A. S. Rosen, S. M. Iyer, D. Ray, Z. Yao, A. Aspuru-Guzik, L. Gagliardi, J. M. Notestein, and R. Q. Snurr
Matter 4, 1578–1597 (2021)

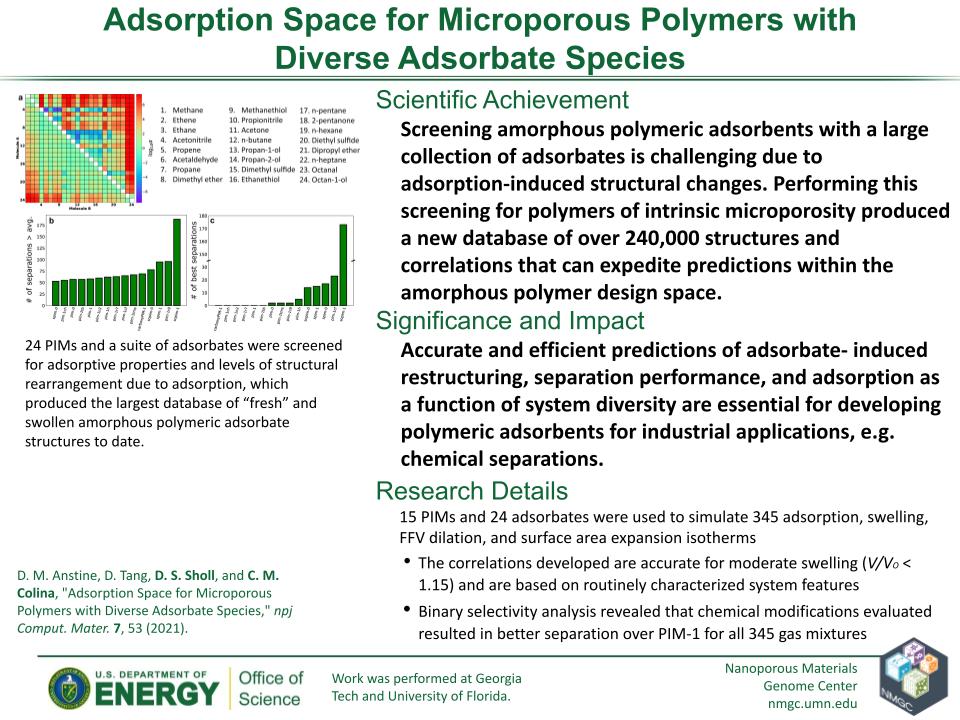
D. M. Anstine, D. Tang, D. S. Sholl, and C. M. Colina
npj Computational Materials 7 (2021)
K. Chen, C. A. Downes, E. Schneider, J. D. Goodpaster, and S. C. Marinescu
ACS Appl. Mater. Interfaces 13, 16384–16395 (2021)

P. Bai, M. Neurock, and J. I. Siepmann
J. Phys. Chem. 125, 6090–6098 (2021)

C. Gu, D. Tang, Z. Yu, J. Liu, and D. S. Sholl
ACS Appl. Mater. Interfaces 13, 11039–11049 (2021)

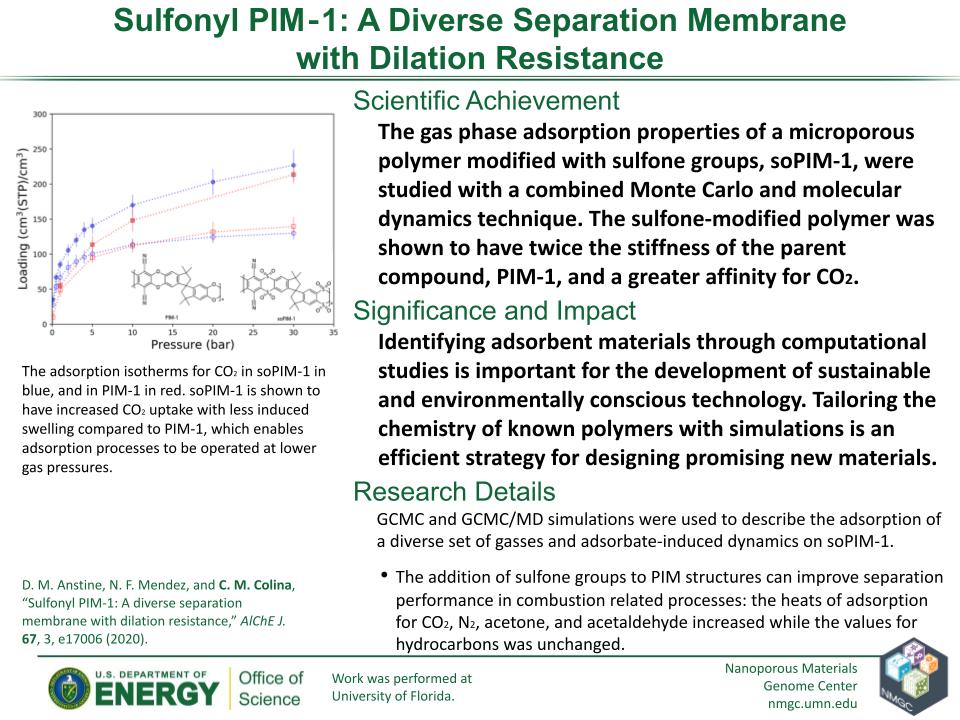
D. M. Anstine, N. F. Mendez, and C. M. Colina
AlChE J. 67, e17006 (2021)
DOI: 10.1002/aic.17006
D. Ray, S. Goswami, J. Duan, J. Hupp, C. Cramer, and L. Gagliardi
Chem. Mater. 33, 1182–1189 (2021)

R. Pollice, G. dos Passos Gomes, M. Aldeghi, R. Hickman, M. Krenn, C. Lavigne, M. Lindner-D'Addario, K. A. Nigam, C.-T. Ser, Z. Yao, and A. Aspuru-Guzik
Acc. Chem. Res. 54, 849–860 (2021)

N. Luo, Z. Hou, C. Zheng, Y. Zhang, A. Stein, S. Huang, and D. G. Truhlar
Chem. Mater. 33, 834–844 (2021)

D. Tang, F. Gharagheizi, and D. S. Sholl
J. Phys. Chem. B 125, 926–936 (2021)

Z. Yao, B. Sánchez-Lengeling, N. S. Bobbitt, B. J. Bucior, S. G. H. Kumar, S. P. Collins, T. Burns, T. K. Woo, O. K. Farha, R. Q. Snurr, and A. Aspuru-Guzik
Nat. Mach. Intell. 3, 76–86 (2021)

T. R. Josephson, P. J. Dauenhauer, M. Tsapatsis, and J. I. Siepmann
J. Comput. Sci. 48, 101267 (2021)

A. Rahbari, T. R. Josephson, Y. Sun, O. A.Moultos, D. Dubbeldam, J. I. Siepmann, and T. J. H. Vlugt
Fluid Phase Equilib. 523, 112785 (2020)

I. Choudhuri and D. G. Truhlar
J. Chem. Theory Comput. 16, 5884–5892 (2020)

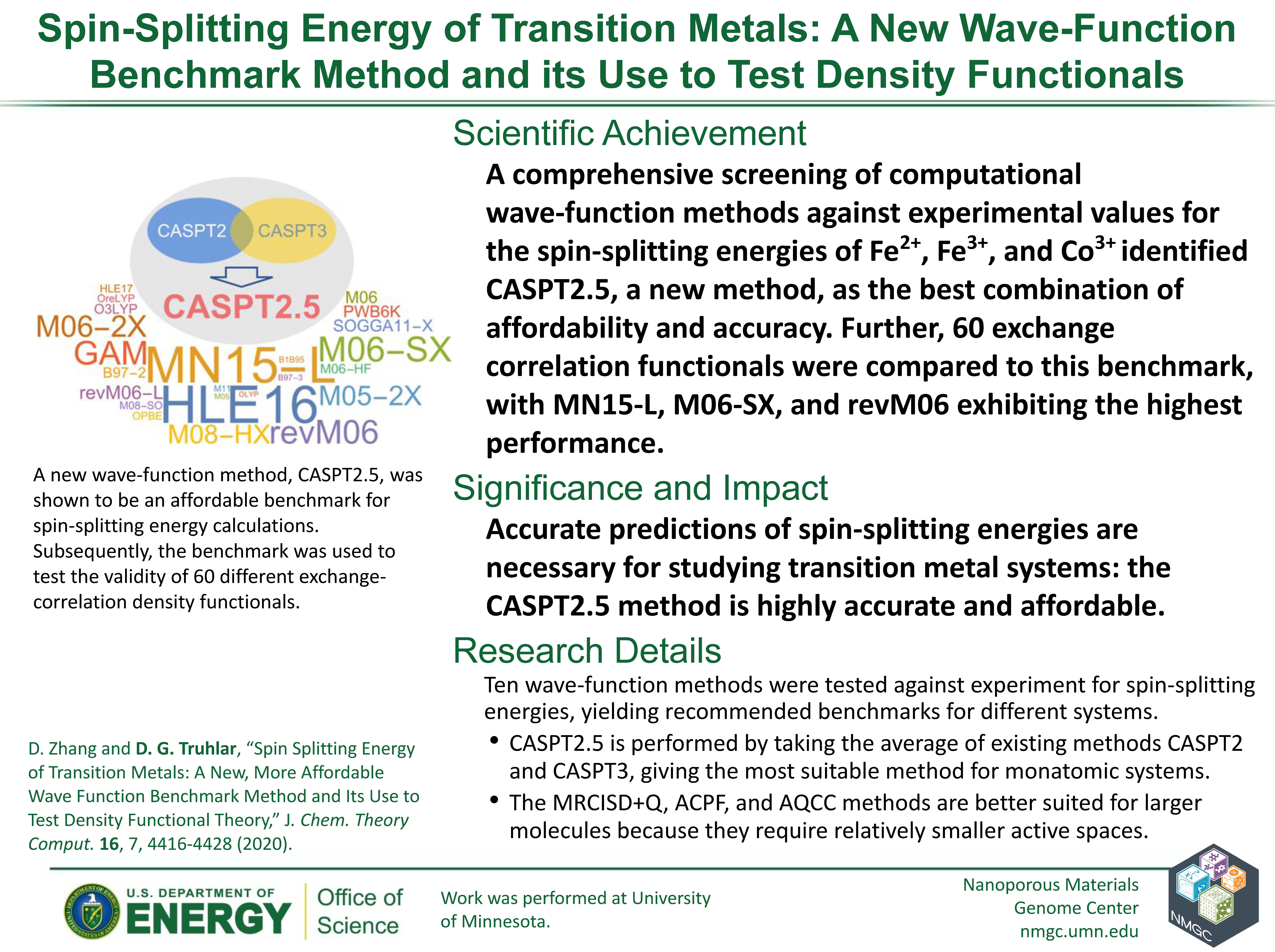
D. Zhang and D. G. Truhlar
J. Chem. Theory Comput. 16, 5432–5440 (2020)

M. L. Mendonca, D. Ray, C. J. Cramer, and R. Q. Snurr
ACS Appl. Mater. Interfaces 12, 35657–35675 (2020)

R. F. DeJaco, K. Loprete, K. Pennisi, S. Majumdar, J. I. Siepmann, P. Daoutidis, H. Murnen, and M. Tsapatsis
AlChE J. 66, e16274 (2020)
DOI: 10.1002/aic.16274

S. M. Pratik, L. Gagliardi, and C. J. Cramer
Chem. Mater. 32, 6137–6149 (2020)

S. Yuan, J. L. Bao, N. Wang, X. Zhang, Y. Wang, D. G. Truhlar, and Y. Xia
Chem. Commun. 56, 8257–8260 (2020)
DOI: 10.1039/D0CC02481C

K. Zhang, K. O. Kirlikovali, J. M. Suh, J.-W. Choi, H. W. Jang, R. S. Varma, O. K. Farha, and M. Shokouhimehr
ACS Appl. Energy Mater. 3, 6019–6035 (2020)

Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, J. J. Eriksen, Y. Gao, S. Guo, J. Hermann, M. R. Hermes, K. Koh, P. Koval, S. Lehtola, Z. Li, J. Liu, N. Mardirossian, J. D. McClain, M. Motta, B. Mussard, H. Q. Pham, A. Pulkin, W. Purwanto, P. J. Robinson, E. Ronca, E. R. Sayfutyarova, M. Scheurer, H. F. Schurkus, J. E. T. Smith, C. Sun, S.-N. Sun, S. Upadhyay, L. K. Wagner, X. Wang, A. White, J. D. Whitfield, M. J. Williamson, S. Wouters, J. Yang, J. M. Yu, T. Zhu, T. C. Berkelbach, S. Sharma, A. Y. Sokolov, and G. K.-L. Chan
J. Chem. Phys. 153, 024109 (2020)
DOI: 10.1063/5.0006074

D. Zhang and D. G. Truhlar
J. Chem. Theory Comput. 16, 4416–4428 (2020)
J. Oktawiec, H. Z. H. Jiang, J. G. Vitillo, D. A. Reed, L. E. Darago, B. A. Trump, V. Bernales, H. Li, K. A. Colwell, H. Furukawa, C. M. Brown, L. Gagliardi, and J. R. Long
Nat. Commun. 11, 3087 (2020)

C.-W. Kung, S. Goswami, I. Hod, T. C. Wang, J. Duan, O. K. Farha, and J. T. Hupp
Acc. Chem. Res. 53, 1187–1195 (2020)

C. Zhao, Z. Yao, D. Zhou, L. Jiang, J. Wang, V. Murzin, Y. Lu, X. Bai, A. Aspuru-Guzik, L. Chen, and Y-S. Hu
Adv. Funct. Mater. 30, 1910840 (2020)

I. Choudhuri and D. G. Truhlar
J. Phys. Chem. C 124, 8504–8513 (2020)

W. Jeong, S. J. Stoneburner, D. King, R. Li, A. Walker, R. Lindh, and L. Gagliardi
J. Chem. Theory Comput. 16, 2389–2399 (2020)

D. S. Graham, X. Wen, D. V. Chulhai, and J. D. Goodpaster
J. Chem. Theory Comput. 16, 2284–2295 (2020)

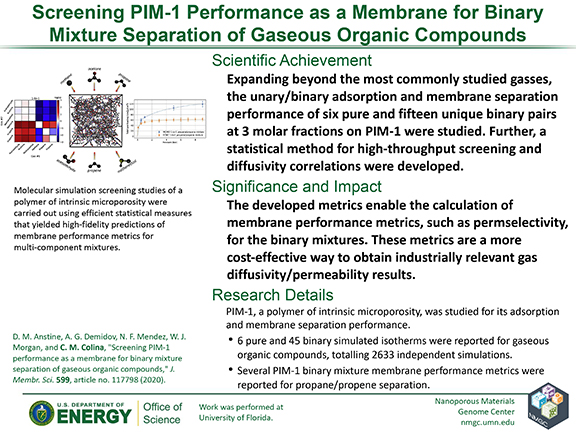
D. M. Anstine, A. G. Demidov, N. F. Mendez, W. J. Morgan, and C. M. Colina
J. Membr. Sci. 599, 117798 (2020)
P. Verma and D. G. Truhlar
Trends Chem. 2, 302–318 (2020)

I. Choudhuri and D. G. Truhlar
Molecules 25, 1552 (2020)

R. F. DeJaco, M. D. de Mello, H. G. T. Nguyen, M. Y. Jeon, R. D. van Zee, M. Tsapatsis, and J. I. Siepmann
AlChE J. 66, e16868 (2020)
DOI: 10.1002/aic.16868

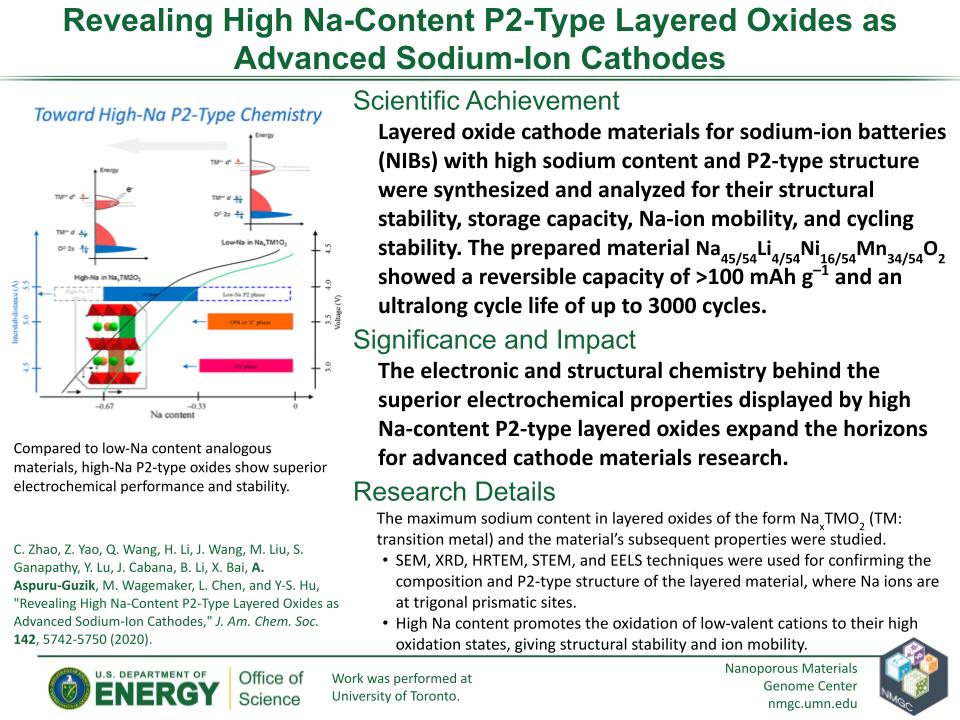
C. Zhao, Z. Yao, Q. Wang, H. Li, J. Wang, M. Liu, S. Ganapathy, Y. Lu, J. Cabana, B. Li, X. Bai, A. Aspuru-Guzik, M. Wagemaker, L. Chen, and Y-S. Hu
J. Am. Chem. Soc. 142, 5742–5750 (2020)
DOI: 10.1021/jacs.9b13572
B. L. Eggimann, Y.-Z.-S. Sun, R. F. DeJaco, R. Singh, M. Ahsan, T. R. Josephson, and J. I. Siepmann
J. Chem. Eng. Data 65, 1330–1344 (2020)

F. Gharagheizi, D. Tang, and D. S. Sholl
J. Phys. Chem. C 124, 3664–3670 (2020)

M. E. Ziebel, C. A. Gaggioli, A. B. Turkiewicz, W. Ryu, L. Gagliardi, and J. R. Long
J. Am. Chem. Soc. 142, 2653–2664 (2020)
DOI: 10.1021/jacs.9b13050

Y. Wang, P. Verma, L. Zhang, Y. Li, Z. Liu, D. G. Truhlar, and X. He
PNAS 117, 2294–2301 (2020)

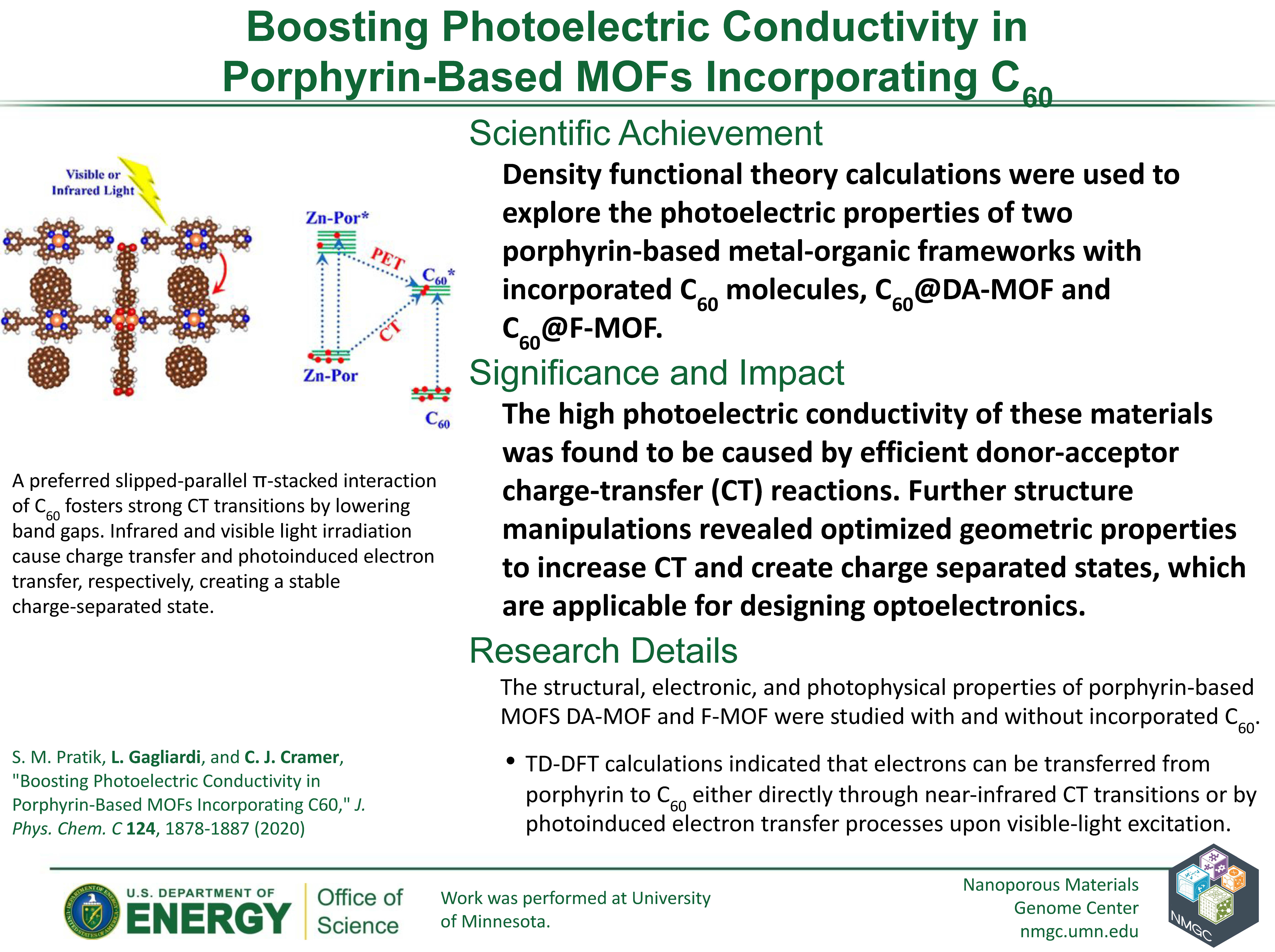
S. M. Pratik, L. Gagliardi, and C. J. Cramer
J. Phys. Chem. C 124, 1878–1887 (2020)
S. Kato, R. J. Drout, and O. K. Farha
Cell Reports Physical Science 1, 100006 (2020)

R. D. Sanner, N. J. Cherepy, H. Q. Pham, and V. G. Young, Jr.
Polyhedron 176, 114256 (2020)

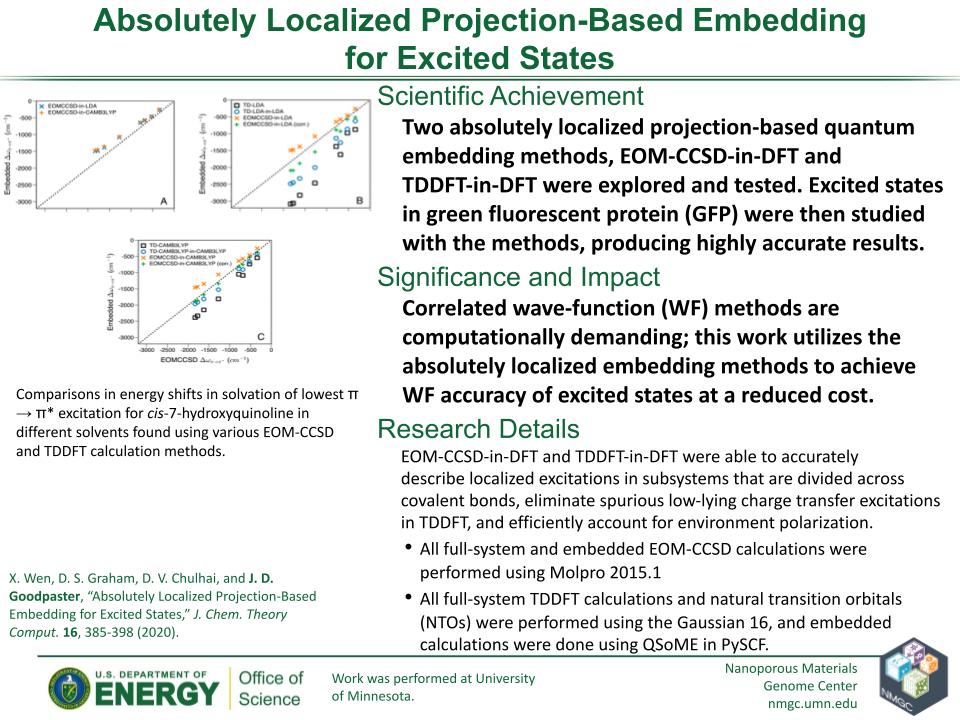
X. Wen, D. S. Graham, D. V. Chulhai, and J. D. Goodpaster
J. Chem. Theory Comput. 16, 385–398 (2020)
H. Q. Pham, M. R. Hermes, and L. Gagliardi
J. Chem. Theory Comput. 16, 130–140 (2020)

Z. Ma, Z. Yao, Y. Cheng, X. Zhang, B. Guo, Y. Lyu, P. Wang, Q. Li, H. Wang, A. Nie, and A. Aspuru-Guzik
Nano Energy 67, 104276 (2020)

X.-P. Wu and D. G. Truhlar
in Computational Photocatalysis: Modeling of Photophysics and Photochemistry at Interfaces, edited by D. Kilin, S. Kilina, and Y. Han (American Chemical Society Symposium Series, Washington, DC, 2019, Chapter 14, 309-326) , ()
DOI: 10.1021/bk-2019-1331

Y. Chung, E. Haldoupis, B. Bucior, M. Haranczyk, S. Lee, H. Zhang, K. Vogiatzis, M. Milisavljevic, S. Ling, J. Camp, B. Slater, J. I. Siepmann, D. S. Sholl, and R. Q. Snurr
J. Chem. Eng. Data 64, 5985–5998 (2019)

A. Gopalan, B. J. Bucior, N. S. Bobbitt, and R. Q. Snurr
Mol. Phys. 117, 3683–3694 (2019)

T. R. Josephson, R. Singh, M. S. Minkara, E. O. Fetisov, and J. I. Siepmann
Mol. Phys. 117, 3589–3602 (2019)

H. Demir, C. J. Cramer, and J. I. Siepmann
Mol. Syst. Des. Eng. 4, 1125–1135 (2019)
DOI: 10.1039/C9ME00095J

X.-P. Wu, I. Choudhuri, and D. G. Truhlar
Energy Environ. Mater. 2, 251–263 (2019)
DOI: 10.1002/eem2.12051

H. Chen, Z. Chen, O. K. Farha, and R. Q. Snurr
ACS Sustainable Chem. Eng. 7, 18242–18246 (2019)

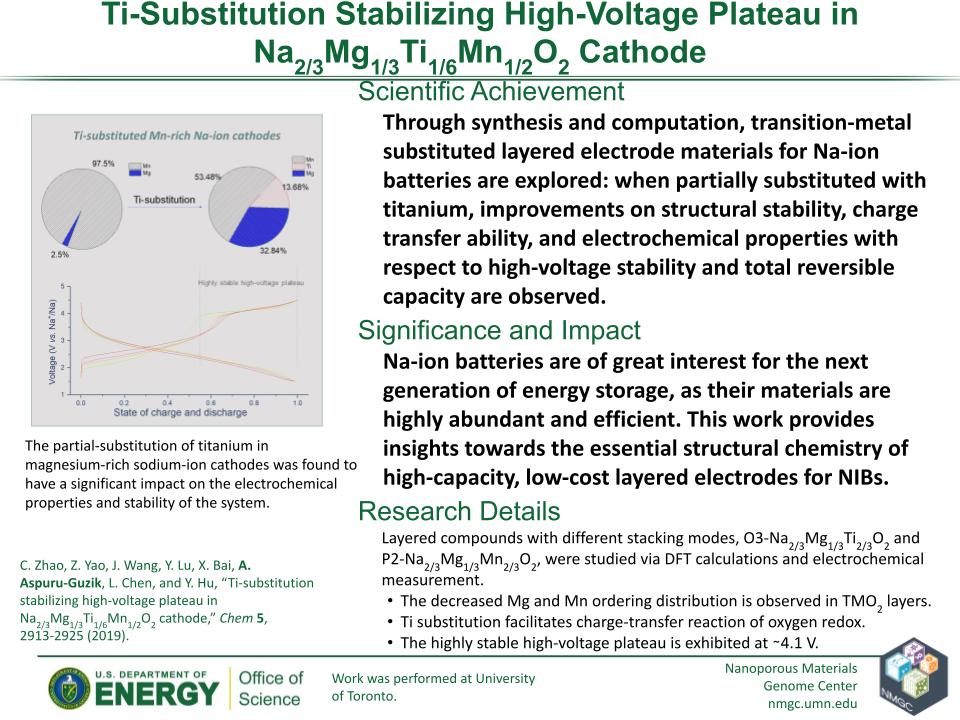
C. Zhao, Z. Yao, J. Wang, Y. Lu, X. Bai, A. Aspuru-Guzik, L. Chen, and Y. Hu
Chem 5, 2913–2925 (2019)
B. J. Bucior, A. S. Rosen, M. Haranczyk, Z. Yao, M. E. Ziebel, O. K. Farha, J. T. Hupp, and J. I. Siepmann, A. Aspuru-Guzik, and R. Q. Snurr
Cryst. Growth Des. 19, 6682–6697 (2019)

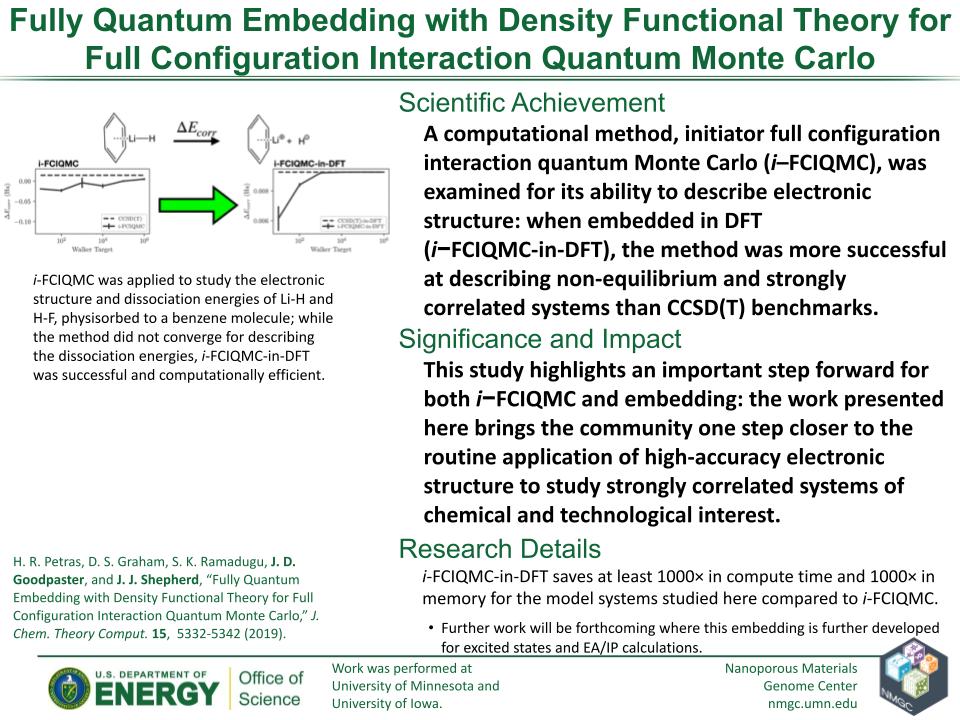
H. R. Petras, D. S. Graham, S. K. Ramadugu, J. D. Goodpaster, and J. J. Shepherd
J. Chem. Theory Comput. 15, 5332–5342 (2019)
R. D. Sanner, N. J. Cherepy, H. P. Martinez, H. Q. Pham, and V. G. Young Jr.
Inorg. Chim. Acta 496, 119040 (2019)

R. Pandharkar, M. R. Hermes, C. J. Cramer, and L. Gagliardi
J. Phys. Chem. Lett. 10, 5507–5513 (2019)

P. Verma, B. G. Janesko, Y. Wang, X. He, G. Scalmani, M. J. Frisch, and D. G. Truhlar
J. Chem. Theory Comput. 15, 4804–4815 (2019)

S. Yuan, J. L. Bao, J. Wei, Y. Xia, D. G. Truhlar, and Y. Wang
Energy Environ. Sci. 12, 2741–2750 (2019)
DOI: 10.1039/C9EE01473J

Q. He, X. Liao, L. Xia, Z. Li, H. Wang, Y. Zhao, and D. G. Truhlar
J. Phys. Chem. C 123, 20737–20747 (2019)

L. G. Gao, R. M. Zhang, X. Xu, and D. G. Truhlar
J. Am. Chem. Soc. 141, 13635–13642 (2019)
DOI: 10.1021/jacs.9b06506

S. M. Pratik and C. J. Cramer
J. Phys. Chem. 123, 19778–19785 (2019)

D. G. Truhlar
J. Chem. Educ. 96, 1671–1675 (2019)

X. Wang, Z. Yao, S. Hwang, Y. Pan, H. Dong, M. Fu, N. Li, K. Sun, H. Gan, Y. Yao, A. Aspuru-Guzik, Q. Xu, and D. Su
ACS Nano 13, 9421–9430 (2019)

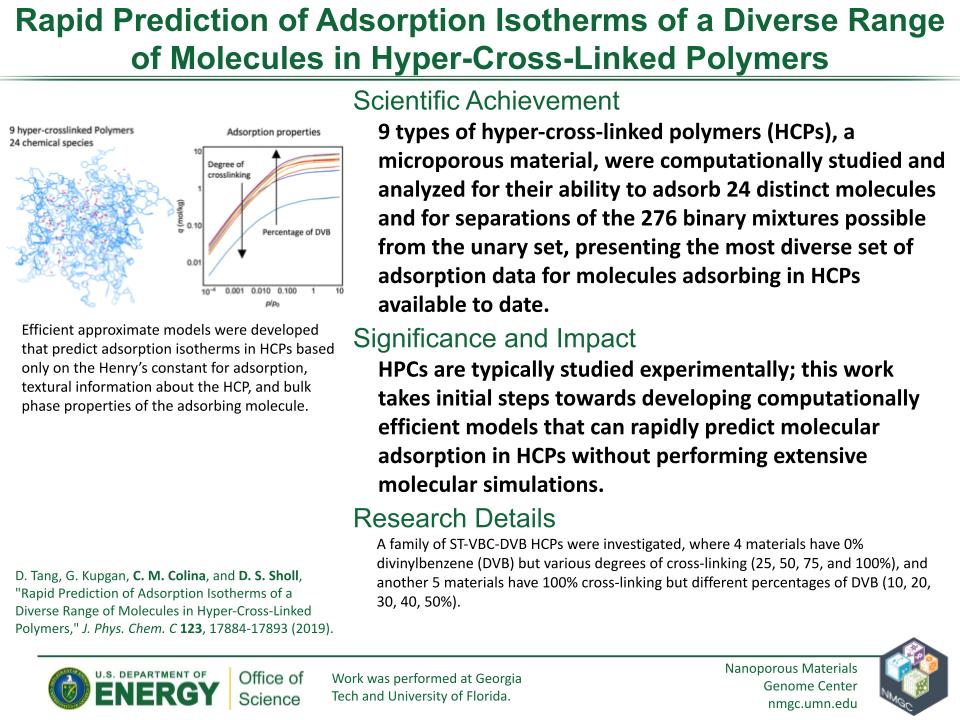
D. Tang, G. Kupgan, C. M. Colina, and D. S. Sholl
J. Phys. Chem. C 123, 17884–17893 (2019)
I. Choudhuri and D. G. Truhlar
J. Phys. Chem. C 123, 17416–17424 (2019)

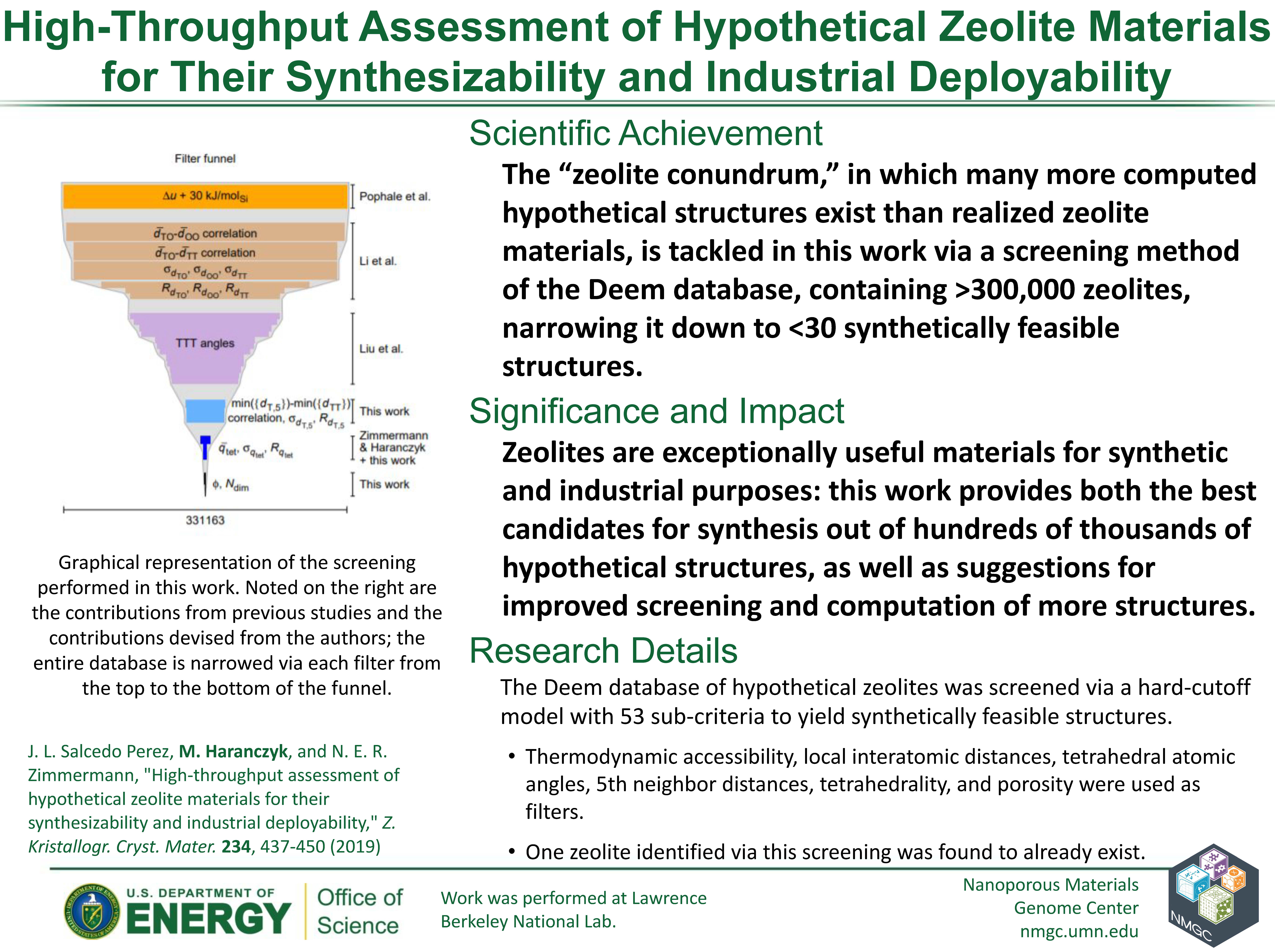
J. L. Salcedo Perez, M. Haranczyk, and N. E. R. Zimmermann
Z. Kristallogr. Cryst. Mater. 234, 437–450 (2019)
R. J. Drout, L. Robison, Z. Chen, T. Islamoglu, and O. K. Farha
Trends in Chemistry 1, 304–317 (2019)

H. Demir, S. Stoneburner, W. Jeong, D. Ray, X. Zhang, O. K. Farha, C. J. Cramer, J. I. Siepmann, and L. Gagliardi
J. Phys. Chem. C 123, 12935–12946 (2019)

Y. Sun, R. F. DeJaco, and J. I. Siepmann
Chem. Sci. 10, 4377–4388 (2019)
DOI: 10.1039/C8SC05340E

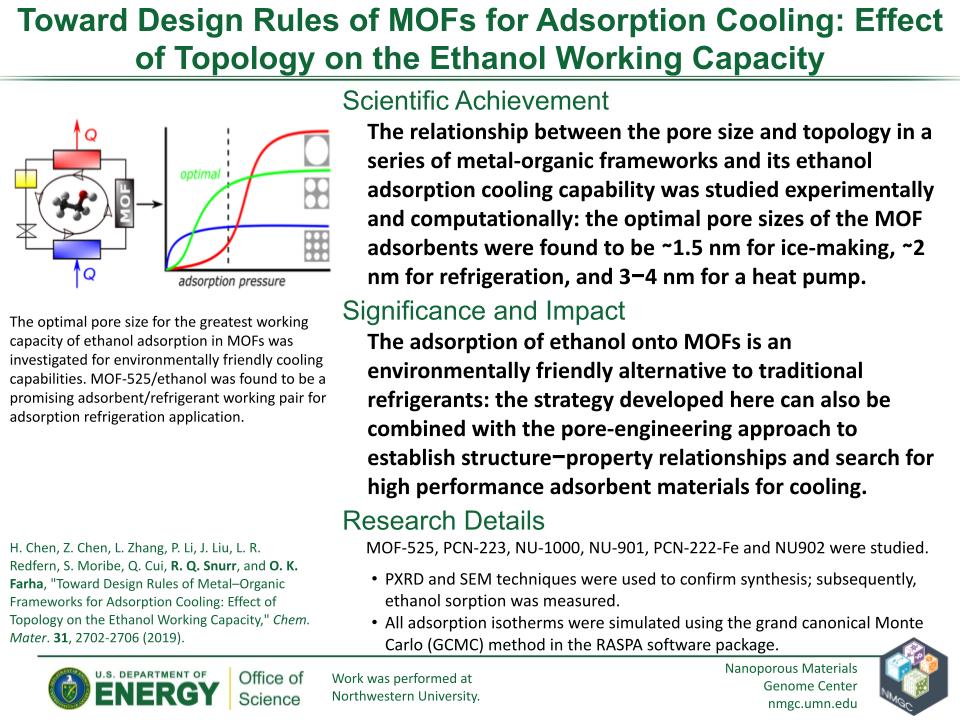
H. Chen, Z. Chen, L. Zhang, P. Li, J. Liu, L. R. Redfern, S. Moribe, Q. Cui, R. Q. Snurr, and O. K. Farha
Chem. Mater. 31, 2702–2706 (2019)
R. J. Verploegh, A. Kulkarni, S. E. Boulfelfel, J. C. Haydak, D. Tang, and D. S. Sholl
J. Phys. Chem. C 123, 9153–9167 (2019)

P. Verma, Y. Wang, S. Ghosh, X. He, and D. G. Truhlar
J. Phys. Chem. A 123, 2966–2990 (2019)

M. L. Aubrey, M. T. Kapelewski, J. F. Melville, J. Oktawiec, D. Presti, L. Gagliardi, and J. R. Long
J. Am. Chem. Soc. 141, 5005–5013 (2019)
DOI: 10.1021/jacs.9b00654

S. Yuan, J. L. Bao, C. Li, Y. Xia, D. G. Truhlar, and Y. Wang
ACS Appl. Mater. Interfaces 11, 10616–10623 (2019)

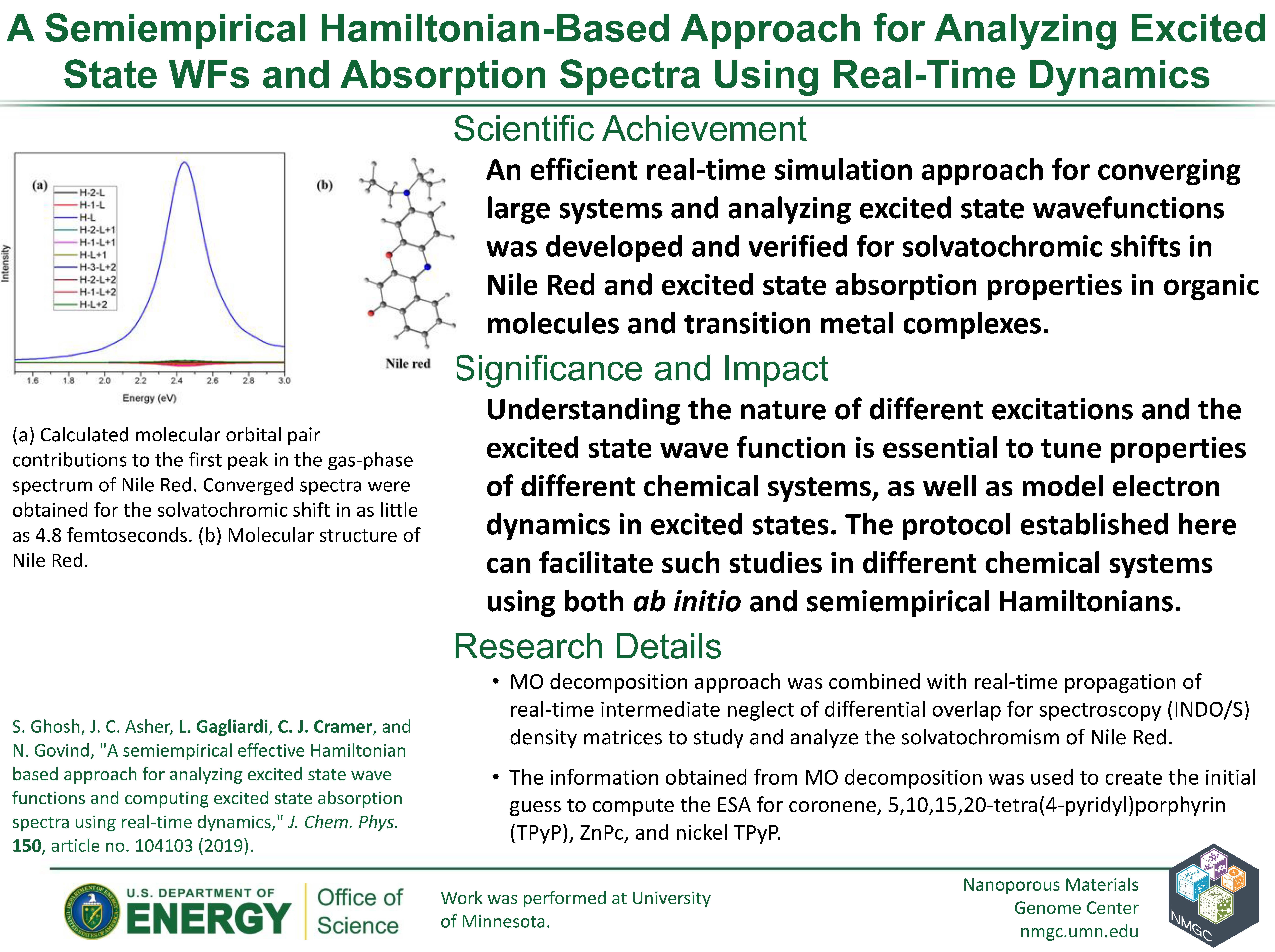
S. Ghosh, J. C. Asher, L. Gagliardi, C. J. Cramer, and N. Govind
J. Chem. Phys. 150, 104103 (2019)
DOI: 10.1063/1.5061746
Z. Yao, V. I. Hegde, and A. Aspuru-Guzik
Adv. Energy Mater. 9, 1802994 (2019)

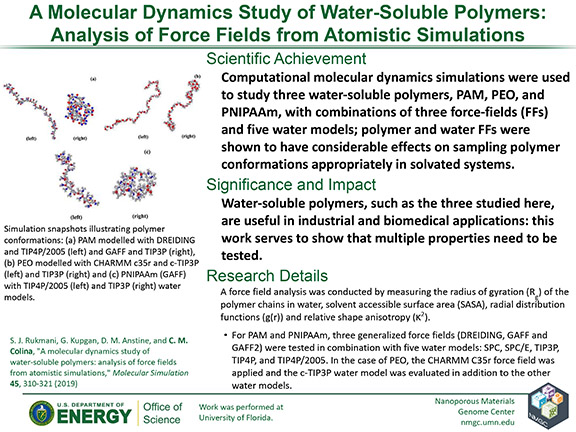
S. J. Rukmani, G. Kupgan, D. M. Anstine, and C. M. Colina
Molecular Simulation 45, 310–321 (2019)
S. Chiniforoush and C. J. Cramer
J. Org. Chem. 84, 2148–2157 (2019)

I. Akpinar, R. J. Drout, T. Islamoglu, S. Kato, J. Lyu, and O. K. Farha
ACS Appl. Mater. Interfaces 11, 6097–6103 (2019)

S. Kato, K.-i. Otake, H. Chen, I. Akpinar, C. T. Buru, T. Islamoglu, R. Q. Snurr, and O. K. Farha
J. Am. Chem. Soc. 141, 2568–2576 (2019)
DOI: 10.1021/jacs.8b12525

M. R. Hermes and L. Gagliardi
J. Chem. Theory Comput. 15, 972–986 (2019)

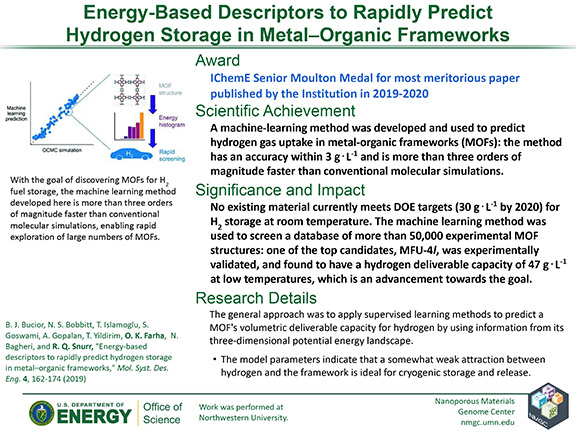
B. J. Bucior, N. S. Bobbitt, T. Islamoglu, S. Goswami, A. Gopalan, T. Yildirim, O. K. Farha, N. Bagheri, and R. Q. Snurr
Mol. Syst. Des. Eng. 4, 162–174 (2019)
DOI: 10.1039/C8ME00050F
X.-P. Wu, L. Gagliardi, and D. G. Truhlar
J. Chem. Phys. 150, 041701 (2019)
DOI: 10.1063/1.5043538

Z. Varga, P. Verma, and D. G. Truhlar
J. Phys. Chem. A 123, 301–312 (2019)

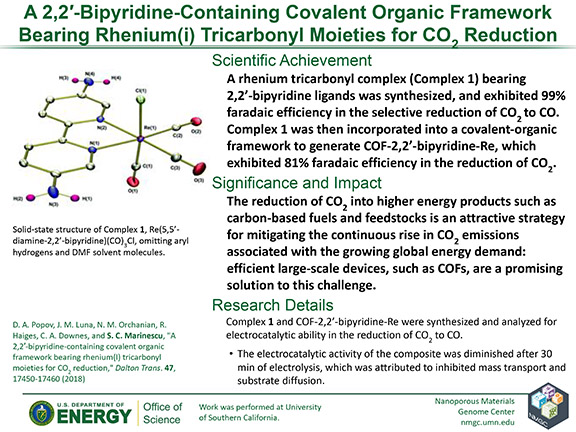
D. A. Popov, J. M. Luna, N. M. Orchanian, R. Haiges, C. A. Downes, and S. C. Marinescu
Dalton Trans. 47, 17450–17460 (2018)
DOI: 10.1039/C8DT00125A
W. You, Y. Liu, J. D. Howe, D. Tang, and D. S. Sholl
J. Phys. Chem. C 122, 27486–27494 (2018)

E. M. Johnson, R. Haiges, and S. C. Marinescu
ACS Appl. Mater. Interfaces 10, 37919–37927 (2018)

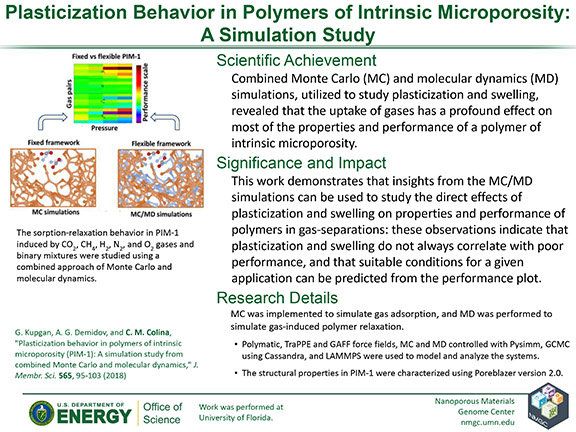
G. Kupgan, A. G. Demidov, and C. M. Colina
J. Membr. Sci. 565, 95–103 (2018)
P. M. Usov, C. F. Leong, B. Chan, M. Hayashi, H. Kitagawa, J. J. Sutton, K. C. Gordon, I. Hod, O. K. Farha, J. T. Hupp, M. Addicoat, A. B. Kuc, T. Heine, and D. M. D’Alessandro
Phys. Chem. Chem. Phys. 20, 25772–25779 (2018)
DOI: 10.1039/C8CP04157A

E. Engelage, N. Schulz, F. Heinen, S. M. Huber, D. G. Truhlar, and C. J. Cramer
Chem. Eur. J. 24, 15983–15987 (2018)

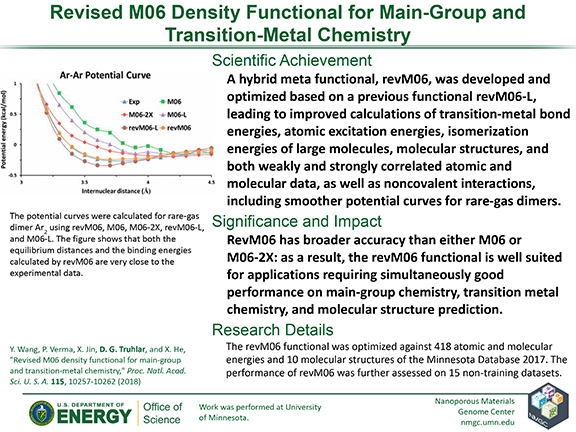
Y. Wang, P. Verma, X. Jin, D. G. Truhlar, and X. He
Proc. Natl. Acad. Sci. U. S. A. 115, 10257–10262 (2018)
E. O. Fetisov, M. Shah, J. R. Long, M. Tsapatsis, and J. I. Siepmann
Chem. Commun. 54, 10816–108192 (2018)
DOI: 10.1039/C8CC06178E

S. J. Stoneburner and L. Gagliardi
J. Phys. Chem. C 122, 22345–22351 (2018)

S. Ghosh, P. Verma, C. J. Cramer, L. Gagliardi, and D. G. Truhlar
Chem. Rev. 118, 7249–7292 (2018)

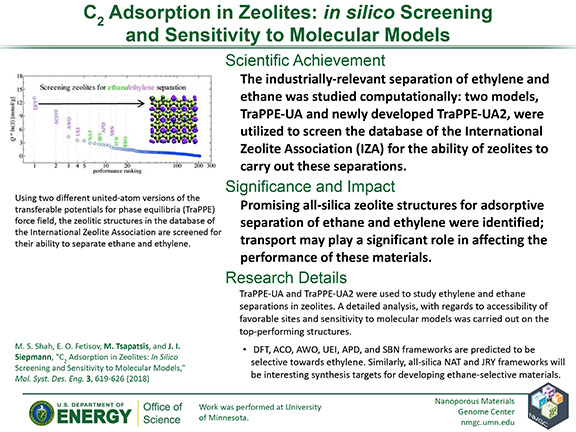
M. S. Shah, E. O. Fetisov, M. Tsapatsis, and J. I. Siepmann
Mol. Syst. Des. Eng. 3, 619–626 (2018)
DOI: 10.1039/C8ME00004B
J. G. Park, M. L. Aubrey, J. Oktawiec, K. Chakarawet, L. E. Darago, F. Grandjean, G. J. Long, and J. R. Long
J. Am. Chem. Soc. 140, 8526–8534 (2018)
DOI: 10.1021/jacs.8b03696

M. R. Momeni and C. J. Cramer
Chem. Mater. 30, 4432–4439 (2018)

X.-P. Wu, L. Gagliardi, and D. G. Truhlar
J. Am. Chem. Soc. 140, 7904–7912 (2018)
DOI: 10.1021/jacs.8b03613

G. Kupgan, L. J. Abbott, K. E. Hart, and C. M. Colina
Chem. Rev. 118, 5488–5538 (2018)

X. Gong, J. Zhou, K. J. Hartlieb, C. Miller, P. Li, O. K. Farha, J. T. Hupp, R. M. Young, M. R. Wasielewski, and J. F. Stoddart
J. Am. Chem. Soc. 140, 6540–6544 (2018)
DOI: 10.1021/jacs.8b03407

S. Goswami, D. Ray, K.-I. Kung, S. Garibay, T. Islamoglu, A. Atilgan, Y. Cui, C. J. Cramer, O. K. Farha, and J. T. Hupp
Chem. Sci. 9, 4477–4482 (2018)
DOI: 10.1039/C8SC00961A

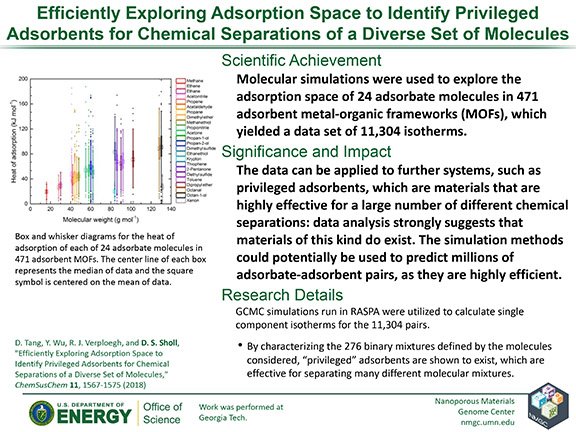
D. Tang, Y. Wu, R. J. Verploegh, and D. S. Sholl
ChemSusChem 11, 1567–1575 (2018)
S. Goswami, J. N. Nelson, T. Islamoglu, Y.-L. Wu, O. K. Farha, and M. R. Wasielewski
Chem. Mater. 30, 2488–2492 (2018)

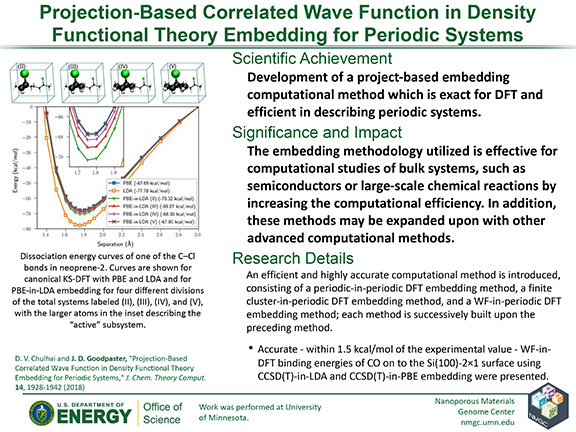
D. V. Chulhai and J. D. Goodpaster
J. Chem. Theory Comput. 14, 1928–1942 (2018)
H. Q. Pham, V. Bernales, and L. Gagliardi
J. Chem. Theory Comput. 14, 1960–1968 (2018)

Y.-L. Wu, N. S. Bobbitt, J. L. Logsdon, N. Powers-Riggs, J. N. Nelson, X. Liu, T. C. Wang, R. Q. Snurr, J. T. Hupp, O. K. Farha, M. C. Hersam, and M. R. Wasielewski
Angew. Chem. Int. Ed. 57, 3985–3989 (2018)

S. Hwang, A. Gopalan, M. Hovestadt, F. Piepenbreier, C. Chmelik, M. Hartmann, R. Q. Snurr, and J. Kärger
Molecules 23 (2018)

P. Verma, Z. Varga, and D. G. Truhlar
J. Phys. Chem. A 122, 2563–2579 (2018)

M. E. Ziebel, L. E. Darago, and J. R. Long
J. Am. Chem. Soc. 140, 3040–3051 (2018)
DOI: 10.1021/jacs.7b13510

E. O. Fetisov, M. S. Shah, C. Knight, M. Tsapatsis, and J. I. Siepmann
ChemPhysChem 19, 512–518 (2018)

T. Islamoglu, M. A. Ortuño, E. Proussaloglou, A. J. Howarth, N. A. Vermeulen, A. Atilgan, C. J. Cramer, and O. K. Farha
Angew. Chem. Int. Ed. 57, 1949–1953 (2018)

C. A. Downes and S. C. Marinescu
ChemSusChem 10, 4374–4392 (2017)

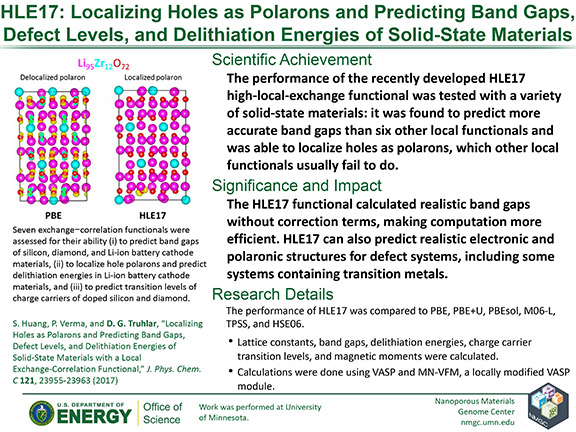
S. Huang, P. Verma, and D. G. Truhlar
Journal of Physical Chemistry C 121, 23955–23963 (2017)
N. Mittal, P. Bai, J. I. Siepmann, P. Daoutidis, and M. Tsapatsis
J. Membr. Sci. 540, 1873–3123 (2017)

T. Zhou, P. Bai, J. I. Siepmann, and A. E. Clark
J. Phys. Chem. C 121, 22015–22024 (2017)
