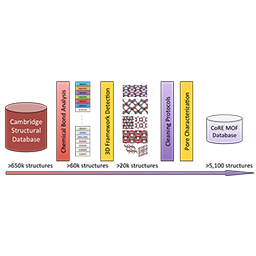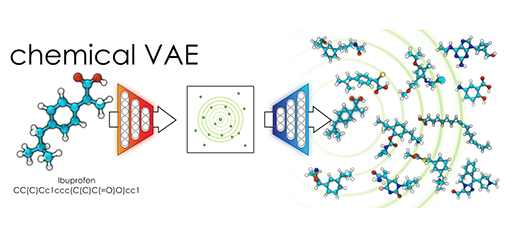
chemical_VAE
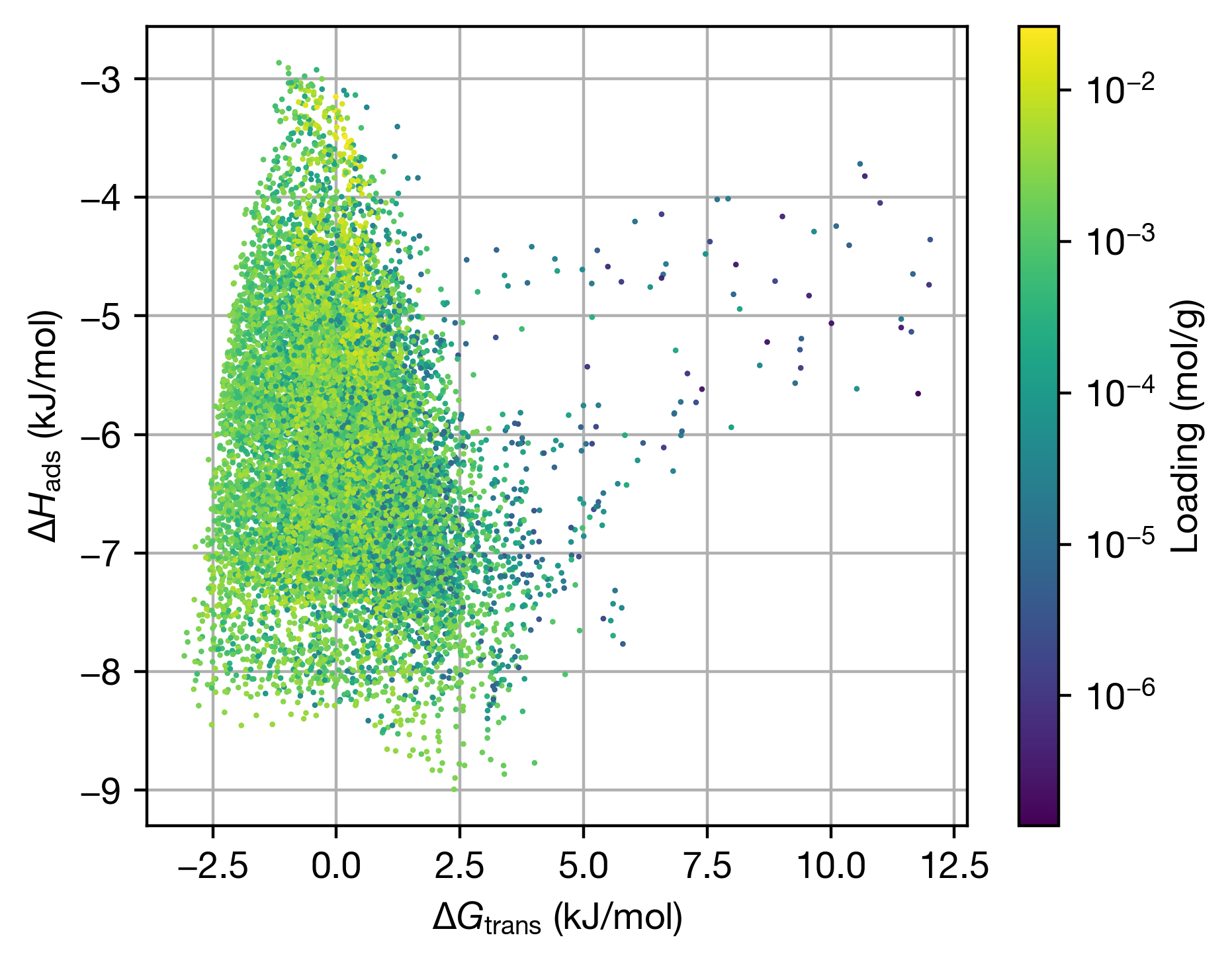
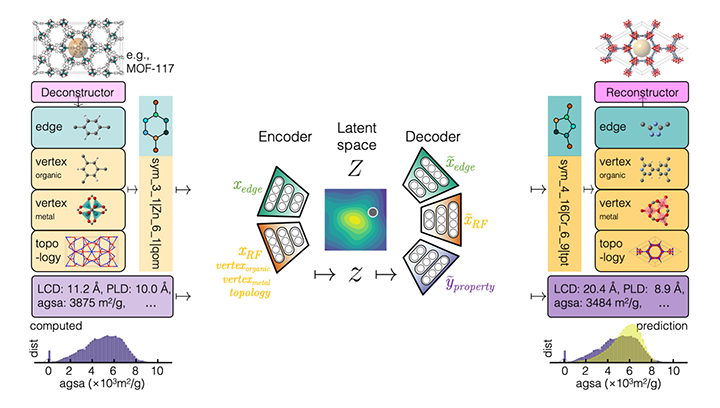
Chemical Variational Autoencoder (chemical_VAE) is a free, open-source software for machine learning of molecular properties. chemical_VAE utilizes molecular SMILES that are encoded into a code vector representation and can be decoded from the code representation back to molecular SMILES. The autoencoder may also be jointly trained with property prediction to help shape the latent space. The new latent space can then be optimized upon to find the molecules with the most optimized properties of interest. chemical_VAE is currently being extended in conjunction with MOFid to capture adsorption of molecules in porous materials.
chemical_VAE can be downloaded from: https://github.com/aspuru-guzik-group/chemical_vae
R. Gómez-Bombarelli, J. Wei, D. Duvenaud, J. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. Hirzel, R. Adams, and A. Aspuru-Guzik, "Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules," ACS Cent. Sci. 4, 268-276 (2018). DOI: 10.1021/acscentsci.7b00572